

Last Updated on October 11, 2025 by Muhamed Elmesery
Polymerase chain reaction (PCR) is now a common and often indispensable technique used in medical and biological research labs for a variety of applications. These include DNA cloning for sequencing, DNA-based phylogeny, or functional analysis of genes, the diagnosis of hereditary diseases, and the detection and diagnosis of infectious diseases.
The wide range of applications of PCR has led to an ever-growing list of variants of the technique. We will focus on the conventional PCR which is the most basic type of PCR reaction and this question: what are the three basic steps of conventional PCR.
Try Conventional PCR in Praxilabs Virtual Labs
Table of Contents
About PCR
This technology is developed by Kary Mullis. In 1993, Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR.
In 1989, Science magazine selected PCR as the “major scientific development” and Taq polymerase, the enzyme essential to PCR’s success, as “molecule of the year.” The advent of PCR meant that insufficiencies in the quantity of DNA were no longer a limitation in molecular biology research or diagnostic procedures. It is indeed difficult to find publications in the biological sciences that do not describe the application of PCR in some or other way.

What Is PCR (polymerase chain reaction)?
The polymerase chain reaction (PCR) is a test tube system for DNA replication which allows a “target” DNA sequence to be selectively amplified several million folds in just a few hours. The PCR achieves amplification of a predetermined fragment of DNA, (the target; which can, e.g., be from 100 to 1000 bp long)
What Is the Conventional PCR Principle?
The concept of DNA amplification by PCR is simple and its impact has been extraordinary.
The chemistry involved in PCR depends on the complementarities (matching) of the nucleotide bases in the double-stranded DNA helix. When a molecule of DNA is sufficiently heated, the hydrogen bonds holding together the double helix are disrupted and the molecule separates or denatures into single strands. If the DNA solution is allowed to cool, the complementary base pairs can reform to restore the original double helix.
What Is the Minimum Data Necessary Before a Typical PCR Reaction Can be Used?
In order to use PCR, the exact sequence of nucleotides that flank (lay on either side of) the area of interest (the target area that needs to be amplified), must be known. This is the absolute minimum data necessary before a typical PCR reaction can be used.
This data is necessary for the design of PCR primers that are 5-3 oligonucleotides of about 20 nucleotides in length. These are designed to be complementary to the flanking sequences of the target area, as mentioned previously. Thus, the researcher has to either use previous data (known information of sequences) or, if this is unavailable, determine the sequence of these regions experimentally. The two primers (primer pair) can then be synthesized chemically and will then serve as leaders or initiators of the replication step.
The key to the replication reaction is that it is driven by a heat-stable polymerase molecule that reads a template DNA in the 3-5 direction and synthesises a new complementary template in the 5-3 direction, using free dideoxy nucleoside triphosphates (dNTP’s = nucleotide bases) as building blocks.
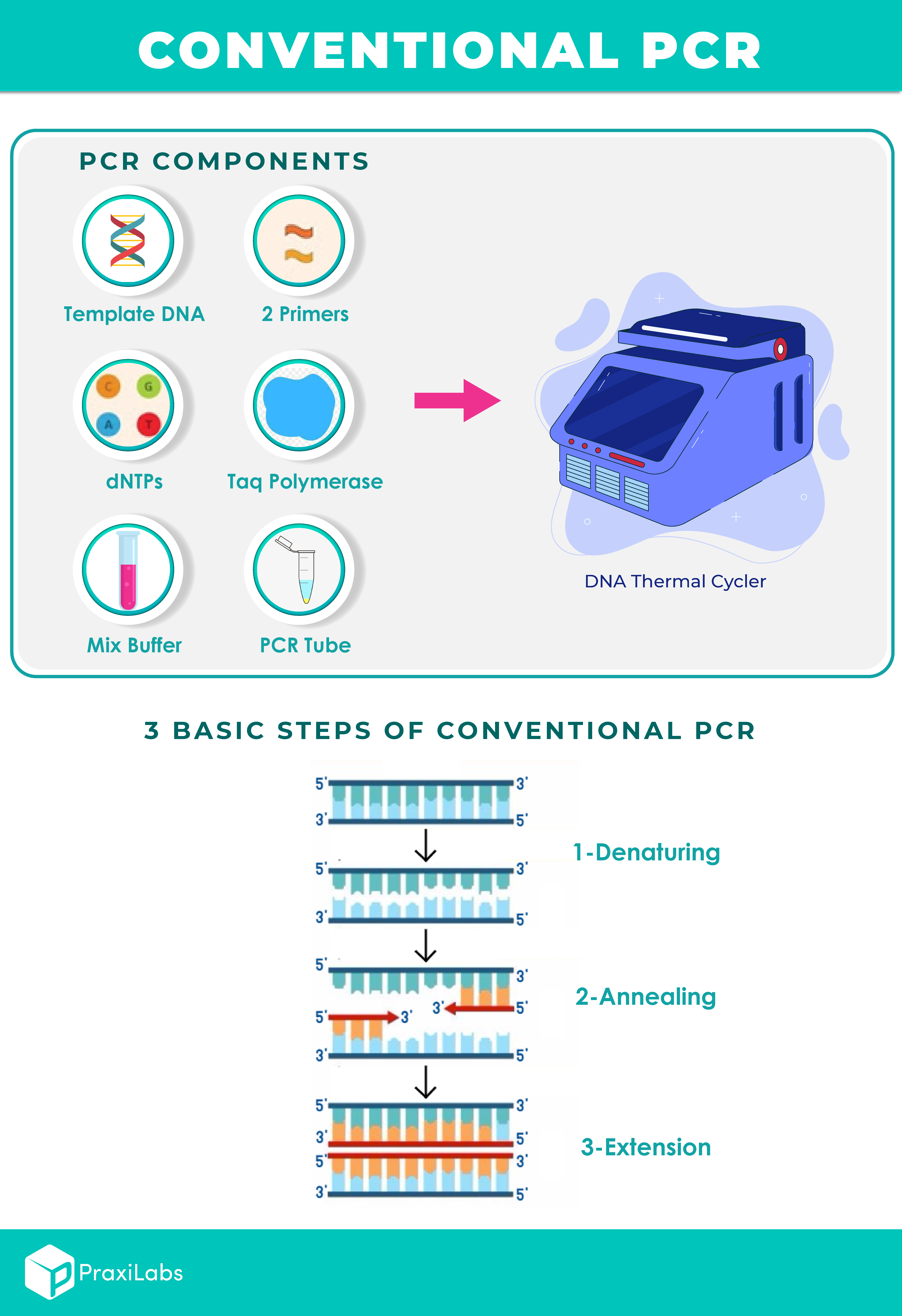
What Are the Components of a Conventional PCR Setup?
- Two primers (Forward and reverse).
- Taq polymerase.
- Deoxynucleoside triphosphates (dNTPs).
- Buffer solution.
- Distilled water.
- Template DNA.
How Does Conventional PCR Work?
PCR is a powerful technique that allows exponential amplification of DNA sequences. A PCR reaction needs a pair of primers that are complementary to the sequence of interest. Primers are extended by the DNA polymerase.
The copies produced after the extension, so called amplicons, are re-amplified with the same primers, thus leading to an exponential amplification of the DNA molecules. After amplification, gel electrophoresis is used to analyse the amplified PCR products and this makes conventional PCR time consuming, since the reaction must finish before proceeding with the post-PCR analysis.
Real Time PCR overcomes this problem, because of its ability to measure the PCR amplicons at early states of the reaction as they are accumulate in a “Real Time Detection” mode, thus measuring the amount of PCR product where the reaction is still in the exponential phase (QPCR).
Try Conventional PCR in Praxilabs Virtual Labs
What Are the Three Basic Steps of Conventional PCR? |PCR Protocol
PCR is based on three simple steps required for any DNA synthesis reaction:
(1) Denaturation of the template into single strands.
(2) Annealing of primers to each original strand for new strand synthesis.
(3) Extension of the new DNA strands from the primers.
These reactions may be carried out with any DNA polymerase and result in the synthesis of defined portions of the original DNA sequence. However, in order to achieve more than one round of synthesis, the templates must again be denatured, which requires temperatures well above those that inactivate most enzymes. Therefore, initial attempts at cyclic DNA synthesis were carried out by adding fresh polymerase after each denaturation step (1,2). The cost of such a protocol becomes rapidly prohibitive.
Typically, PCR consists of a series of 20–40 repeated temperature changes, called cycles, with each cycle commonly consisting of 2–3 discrete temperature steps. The cycling is often preceded by a single temperature step at a high temperature (>90 °C) and followed by one hold at the end for final product extension or brief storage. The temperatures used and the length of time applied in each cycle depend on a variety of parameters. These include the enzyme used for DNA synthesis, the concentration of divalent ions and dNTPs in the reaction, and the melting temperature of the primers.
Flowchart of the three main steps of PCR
What are the performing steps of PCR?
Now we will know What happens at each stage of PCR
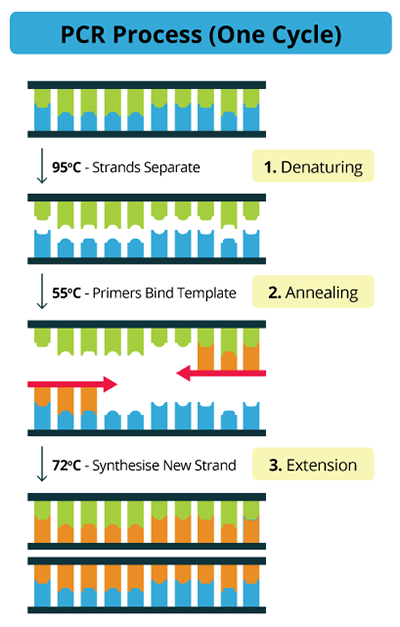
Initialization step
(only required for DNA polymerases that require heat activation by hot-start PCR): This step consists of heating the reaction to a temperature of 94–96 °C (or 98 °C if extremely thermostable polymerases are used), which is held for 1–9 minutes.
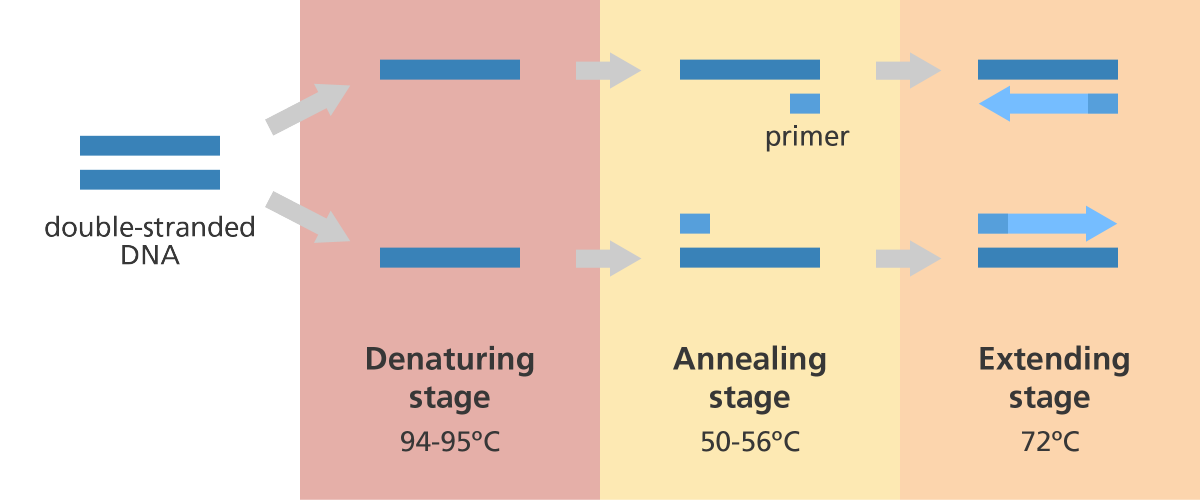
Denaturation step
This step is the first regular cycling event and consists of heating the reaction to 94–98 °C for 20–30 seconds. It causes DNA melting of the DNA template by disrupting the hydrogen bonds between complementary bases, yielding single-stranded DNA molecules.
Annealing step
The reaction temperature is lowered to 50–65 °C for 20–40 seconds allowing annealing of the primers to the single-stranded DNA template.
Extension/elongation step
The temperature at this step depends on the DNA polymerase used; Taq polymerase has its optimum activity temperature at 75–80 °C and commonly a temperature of 72 °C is used with this enzyme. At this step, the DNA polymerase synthesizes a new DNA strand complementary to the DNA template strand by adding dNTPs that are complementary to the template in 5′ to 3′ direction, condensing the 5′-phosphate group of the dNTPs with the 3′-hydroxyl group at the end of the nascent (extending) DNA strand. The extension time depends both on the DNA polymerase used and on the length of the DNA fragment to amplify.
3 steps of PCR and temperatures
PraxiLabs 3D virtual biology laboratory provides the PCR experiment, where you can conduct the experiment in the virtual biology lab, which provides science students and professors with a more accurate understanding of PCR meaning.
PCR Design Tool
Designing appropriate primers is essential to the successful outcome of a PCR experiment. When designing a set of primers for a specific region of DNA desired for amplification, one primer should anneal to the plus strand, which by convention is oriented in the 5′ → 3′ direction and the other primer should complement the minus strand, which is oriented in the 3′ → 5′ direction.
Below is a list of characteristics that should be considered when designing primers.
- Primer length should be 15-30 nucleotide residues (bases).
- The Optimal G-C content should range between 40-60%.
- The 3′ end of primers should contain a G or C in order to clamp the primer and prevent “breathing” of ends, increasing priming efficiency. DNA “breathing” occurs when ends do not stay annealed but fray or split apart. The three hydrogen bonds in GC pairs help prevent breathing but also increase the melting temperature of the primers.
- The 3′ ends of a primer set, which includes a plus strand primer and a minus strand primer, should not be complementary to each other, nor should the 3′ end of a single primer be complementary to other sequences in the primer. These two scenarios result in the formation of primer dimers and hairpin loop structures, respectively.
- Optimal melting temperatures (Tm) for primers range between 52-58 °C, although the range can be expanded to 45-65 °C. The final Tm for both primers should differ by no more than 5 °C.
- Di-nucleotide repeats (e.g., GCGCGCGCGC or ATATATATAT) or single base runs (e.g., AAAAA or CCCCC) should be avoided as they can cause slipping along the primed segment of DNA and or hairpin loop structures to form. If unavoidable due to the nature of the DNA template, then only include repeats or single base runs with a maximum of 4 bases.
Notes: There are many computer programs designed to aid in designing primer pairs, ex: NCBI Primer design tool
Source: Lorenz, T.C. (2012) Polymerase chain reaction: Basic protocol plus troubleshooting and optimization strategies, Journal of visualized experiments : JoVE. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4846334/ (Accessed: 30 July 2024).
Applications of PCR
Because of the great sensitivity, PCR has found popularity in a wide range of applications:
- DNA sequencing and gene reproduction & cloning.
- The cellular cloning is one of the most remarkable applications of PCR. It makes it possible to isolate, that is to say, to purify a gene without resorting to traditional methods of molecular cloning which consist in inserting a DNA library in a plasmid vector which is then used to transform a bacterial strain whose clones after selection are screened. A cellular cloning is used when using PCR because it is useless to use a cellular system (bacteria, yeast, and animal or plant cell) to amplify the clone.
- The identification of genetic fingerprints used in forensic scientists which use PCR to connect blood, saliva, or tissue left at the scene of a crime to a suspect or victim.
- Discovering the possibility of transmitting genetic diseases between potential parents and determining whether the diseases will or will not be passed to their next generation of children.
- DNA-based phylogeny, or functional analysis of genes.
- The diagnosis of hereditary diseases.
- The detection and diagnosis of infectious diseases.
- PCR can be done to investigate locus-specific methylation which is considered as an important biomarker for disease diagnosis (ex: cancers, Parkinson’s disease, obesity, insulin resistance, Alzheimer’s disease, etc)
- In the methylation-specific PCR (MSP) method, two primer pairs are designed to differentiate the methylation state of the locus of interest.
- The DNA samples are first treated with bisulfite to convert unmethylated cytosine (C) to uracil (U).
- Methylated cytosine remains unaffected by the bisulfite treatment.
- To detect the methylated sites, one pair of primers is designed with guanine (G) to pair with methylated cytosine in the target sequence.
- To detect the unmethylated sites, another pair of primers is designed with adenine to pair with uracil in the bisulfite-converted molecules (and then pair with thymine in subsequent PCR cycles).
- Positive PCR amplification resulting from primer binding is used to detect the methylation state of the locus.
PCR and Infectious Diseases
Infectious diseases can be caused by microbial pathogens, including agents of fungal, protozoan, bacterial, clamydial, rickettsia and viral nature. Despite many advances in diagnostics and vaccinology, infectious diseases still have devastating consequences for agricultural economies, worldwide.
Three examples of devastation with regard to animal husbandry since the 1990s include the emergence of the prion, bovine spongiform encephalopathy (BSE); the huge outbreaks of foot-and-mouth disease (FMD) in Europe and avian influenza (AI) in Asia and elsewhere. A great many of these infectious diseases can be transmitted from vertebrate animals to man (called zoonoses) where more than 200 such zoonotic diseases are known.
Infectious diseases are typically transmitted through the skin or eyes (direct contact, insect vectors, bite wounds, and sexual contact). In other cases agents are airborne and infect the epithelial cells lining the respiratory tract from where further systemic infection may proceed. Additional sources of infectious microorganisms are contaminated food and water with a route of infection through the mouth and alimentary tract, or through the respiratory system.
Laboratory diagnostic technology is directed towards either the detection of the presence or absence of a pathogen and its subsequent identification and characterization The detection of the pathological effect of, or immunological response to, infection by a particular pathogen.
PCR represents an entirely new technology. In vitro bacterial or viral culture is widely used to isolate and multiply pathogens, so that the organism itself, or its antigens, can be more readily detected, by being present in greater quantity and generally with fewer contaminants.
PCR technology permits the same principle (i.e., in vitro amplification) to be applied to the detection of specific sequences of nucleic acid. There are enormous benefits to this approach. The application of PCR to disease diagnosis has been somewhat restricted to laboratories with the required facilities, equipment, funding, and expertise. The procedure must be made in very clean conditions since contamination with minute amounts of extraneous DNA may produce false positive results.
Get Started Praxilabs For FREE!
PCR Analysis and COVID-19 Infection Detection
One of the most important applications of PCR now is the detection of COVID-19 Infection.
Recently, with the emergence of COVID-19 virus, we often hear the term “PCR analysis.” Which is the basic analysis currently used to detect Infected people with COVID-19.
Through PCR analysis, scientists can detect the presence of viruses that cause infection, even when they are present in small quantities in the body. This method contributes to the diagnosis of transmissible viral diseases, as well as to the identification of mutations in various genetic disorders.
For more information, write our article PCR Analysis: COVID-19 Infection Detection Method… PraxiLabs Initiative Experiments

PraxiLabs Virtual Lab of Conventional PCR
After performing a conventional PCR simulation using PraxiLabs virtual labs for biology, students gain hands-on experience of the principle and practice of conventional polymerase chain reaction (PCR).
Remember,a PCR experiment should ideally be performed in a dedicated ‘clean’ area which is free from other work involving DNA.
Steps of the Experiment
- Set up the reaction wearing gloves at all times and label the lids of the eppendorf.
- Prepare an amount of the Master Mix sufficient to 6 samples of the DNA by adding the special reagents for the experiment in the master mix tube.
- 243 ml of distilled water (40.5*6).
- 30 ml of the 10X PCR buffer containing MgCl2 (5*6).
- 6 ml of the 10 Mm dNTPs (1*6).
- 6 ml of the Forward primer (1*6).
- 6 ml of the Reverse primer (1*6).
- 3 ml of the Taq DNA polymerase (0.5*6).
- Add 49 ml of the Master Mix you just prepared to 1 ml of each DNA sample.
- Place the eppendorfs in the thermal cycler and carry out an initialization denaturation at 94 degrees Celsius for 5 minutes for one cycle.
- Carry out multiplication cycles, which is divided into three consecutive steps:
- Denaturation step: at 94 degrees Celsius for 30 seconds, 30 cycles.
- Annealing step: at 50-65 degrees Celsius for 30 seconds, 30 cycles.
- Elongation step: at 72 degrees Celsius for one minute, 30 cycles.
- Carry out final elongation at 72 degrees Celsius for 10 minutes for one cycle.
- Carry out the final step in the experiment which is the final hold at 4 degrees Celsius for an indefinite time for one cycle.
Video To Clarify The Steps of Conventional PCR Simulation
Conventional PCR Unveiled: Your FAQs Clarified
What is Polymerase Chain Reaction (PCR)?
Polymerase chain reaction (PCR) is a vital technique developed by Kary Mullis to be used in medical and biological research labs for several purposes. The PCR procedure is used mainly to make millions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. It involves denaturation, annealing, and extension of DNA strands.
The principle of PCR depends on the complementarities of the nucleotide bases in the double-stranded DNA helix. When a molecule of DNA is sufficiently heated, the hydrogen bonds holding together the double helix are disrupted and the molecule separates or denatures into single strands. If the DNA solution is allowed to cool, the complementary base pairs can reform to restore the original double helix.
What are the three basic steps of conventional PCR case one?
The three basic steps of conventional PCR, as we explained previously in our blog, are:
(1) Denaturation of the template into single strands.
(2) Annealing of primers to each original strand for new strand synthesis.
(3) Extension of the new DNA strands from the primers.
These reactions may be carried out with any DNA polymerase and result in the synthesis of defined portions of the original DNA sequence. However, in order to achieve more than one round of synthesis, the templates must again be denatured, which requires temperatures well above those that inactivate most enzymes. Therefore, initial attempts at cyclic DNA synthesis were carried out by adding fresh polymerase after each denaturation step (1,2). The cost of such a protocol quickly becomes prohibitive.
What are the four major steps of PCR in order?
Denaturation of the double-stranded DNA (template) into two single strands by heat (at temperatures higher than 90 degrees Celsius). The Heat breaks down hydrogen bonds between the base pairs, while the stronger bonds between deoxyribose and phosphates remain intact.
Annealing of the primers to each original strand for new strand synthesis, the target DNA is targeted using primers that target that bind to the ends of the target DNA sequence at temperature 40 – 65 degrees Celsius depending on the base and length sequence of the primers.
Extension: the temperature is increased to 72 degrees Celsius and the enzyme “Taq DNA polymerase” is used to replicate the DNA strands. During the extension step, two identical double stranded DNA molecules are synthesized. After the first PCR cycle, the process is generally repeated between 25 and 35 times.
Analysis with gel electrophoresis: we can evaluate the amplified DNA and identify the nucleotide sequences by detecting the bands or ladder-like steps that migrate to the same levels in the gel.




