
Last Updated on October 11, 2025 by Muhamed Elmesery
Western blot is an indispensable mechanism in the modern biomedical research laboratory, as well as in laboratories doing research in other areas.
This technique is routinely used now for research purposes, for instance, clinical medical laboratories.
Western blot can be used to identify a specific protein in a sample. Also, it provides information about protein size and relative abundance in the sample.
Try Western Blot Simulation For FREE
Western blot is sometimes called “protein immunoblot” or “western blotting.”
It is considered as an analytical technique used mainly in molecular biology and immunogenetics where antibodies are used to specifically detect their antigen.
The term “blotting” refers to the transfer of biological samples from a gel to a membrane and their subsequent detection on the surface of the membrane.

The development of the protein blotting techniques is credited to “Harry Towbin.” In 1978 “Harry Towbin” was hired by “Julian Gordon” at the “Friedrich-Miescher-Institute” in Basel for a postdoctoral fellowship.
“Harry” and “Julian” worked in cooperation with “Theo Staehelin,” a pioneer in the field of initiation factors for mammalian protein synthesis who had ample expertise in ribosomes.
In 1979 they published a paper entitled “Electrophoretic Transfer of Proteins From Polyacrylamide Gels to Nitrocellulose Sheets: Procedure and Some Applications.” This paper provides the beginning of the protein blotting techniques.

Try PraxiLabs Virtual Biology Lab For FREE and Enjoy Science Education Anywhere and Anytime
Before discussing the principal and applications of western blotting technique . We need to clarify some definitions similar to western blot such as eastern blot, northern blot, and northwestern blot.
Actually, the eastern blot can be considered as an extension of the biochemical technique of western blot and it is often used to detect carbohydrate epitopes.
On another front, northern blot also called RNA blot is a technique used in molecular biology research to study gene expression by detection of RNA (or isolated mRNA) in a sample.
By combining the northern and western blot, this produced a hybrid called northwestern blot which was used in molecular biology to detect interactions between RNA and proteins.
Through the following lines, we will discuss the western blot technique in detail, its applications, and how to perform it.
Table of Contents
Principle of Work
Western blot is a commonly used technique designed to study a specific protein in a sample containing many other proteins.
Proteins in the sample are extracted then separated by gel electrophoresis (SDS-PAGE) according to their molecular weight. Then, the proteins are electrophoretically transferred (blotted) to a more durable surface (e.g. nitrocellulose or PVDF membrane).
Finally, a specific antibody is used to detect the protein of interest. The bound and labeled protein can be visualized giving information about its molecular weight and relative quantity.
Watch the walkthrough video and get a full understanding of how to conduct the western blot simulation in PraxiLabs’ Virtual Science Labs.
Process of Western Blot Experiment
Introduction
The western blotting method was proposed first by Towbin in 1979 as we mentioned above. The main Idea of the Western blotting as stated is detecting and analyzing proteins.
It relies on the blotting technique which involves transferring proteins after electrophoresis separates them from the gel to a membrane, then they can be ready to be visualized.
After preparing and loading the sample onto a gel, the electrophoresis process starts and the negatively charged proteins move toward the positively charged anode.
After the electrophoresis phase, the protein must be transferred from the gel to a membrane In order to further analyze proteins, the procedure of transferring them onto a membrane is called blotting.
To prevent unwanted membrane-protein interaction, the membrane must be blocked after the transfer. Then the membrane is commonly first probed using a primary protein-specific antibody followed by a labeled secondary antibody used for detection in order to visualize the targeted protein.
The estimation of protein size can be achieved by adding a separate marker solution to one of the wells in the gel. In addition, the antibody interactions are used to verify specific protein.
The size is not the only factor that causes the separation on the gel, the separation could be due to the molecular charge, hydrophobic regions, and degree of denaturation.
The outcome of a western blot technique depends on three important factors:
- The ability of the antibody to bind a specific protein.
- The strength of the interaction.
- The concentration of the protein of interest itself.
Moreover, the specificity of the binding to the target and low cross-reactivity are important features as well.
The results of the western blot are not always easy to interpret as the size of the protein may vary from the theoretical weight due to posttranslational modifications, such as glycosylation, or interactions with other proteins.
However, the western blot is a very common method and almost all available commercial antibodies have been validated using this method.
Preparing Samples
In the first step, samples are prepared and loaded on to a gel. Your sample could be tissue, cells, or other solution, that you want to extract and analyze its protein.

Using blending, homogenization, or sonication, the tissue will be broken down. While the buffers and inhibitor will play their role. Adding buffers will lead to lyse the cells and solubilize the proteins, where the adding of inhibitors will prevent denaturation or degradation.
You can apply various types of filtration and centrifugation methods for further preparation for the samples.
Take in mind that the determination of total protein concentration of the generated extract is important to be able to load a specific amount on the gel to enable comparison between samples.
The extract will then be diluted with a loading buffer consisting of glycerol and a dye. The importance of the glycerol is raising the density to simplify the loading and the dye is used to visualize the sample.
To break the structures of the protein you will apply heat on the samples, which will help to keep the negative charge from neutralization.
It is better to include the positive and negative controls in the set up to confirm the identity of the protein as well as the activity of the antibody.

Electrophoretic Separation of Proteins
Gel electrophoresis is a technique in which charged molecules such as protein are separated according to physical properties as they are forced through a gel by an electrical current.
After your sample is ready to be loaded to separate the proteins according to size by gel electrophoresis. Apply the electric field over the gel to separate the charged molecules.
This method is called polyacrylamide gel electrophoresis (PAGE) which is used to apply the separation step for proteins when using native condition.
PAGE is a powerful analytical tool providing information on the mass, charge, purity, or presence of a protein.
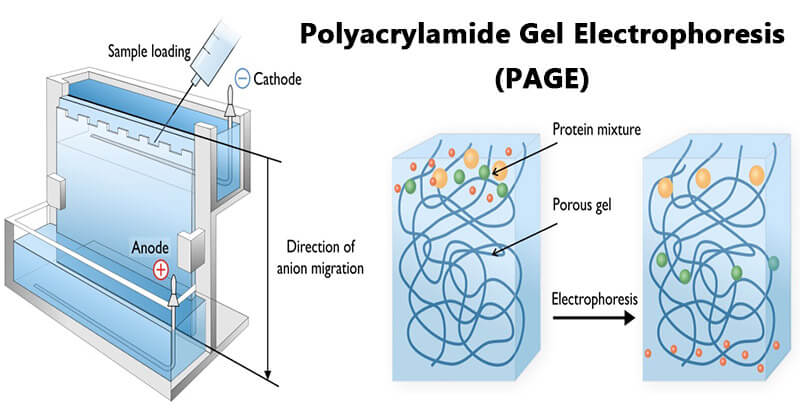
Then sodium dodecyl sulfate (SDS) is added to the system in order to provide the denaturing conditions, so the method is called SDS-PAGE.
The role of the SDS is to bind to the protein to form a micelle with a negative charge around the protein regardless of its inherent charge.
The importance of the denaturing condition can be concluded in the dissolving of the tridimensional structure of the proteins and they gain a negative charge in proportion to their molecular size, then they travel in the acrylamide gel according to their molecular sizes. The smaller the size of the running protein, the faster it travels through the pores of the gel.
At the native conditions, the mobility will depend on both charge and hydrodynamic size allowing detection of changes in charge due to chemical degradation, conformational changes due to folding or unfolding, aggregation, or other binding events.
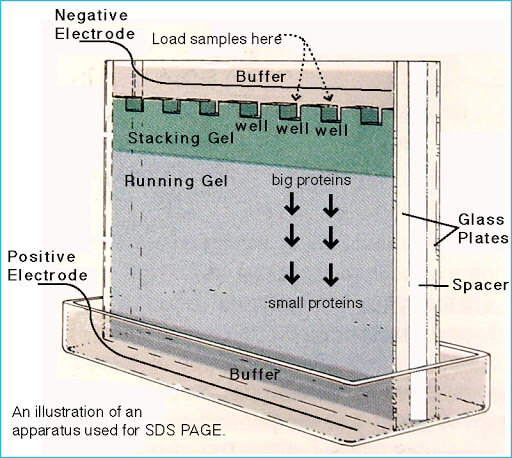
The used gel here is divided into two parts that differ in densities:
- Stacking gel.
- Separating gel.
These two sections have different pH and gel concentration values. On one hand, acidic pH and lower concentration of the stacking gel cause poor separation for the proteins. But it allows them to form highly defined sharp bands before they enter the separating gel.
On the other hand, the basic conditions and higher gel concentration of the separating gel make proteins differentiate by size, as smaller proteins travel faster in the gel than bigger ones.
Precast gels are convenient; however, it is possible to cast them by hand.
The gel is immersed in the buffer and the protein samples and markers are loaded to the wells in the gel. Then by applying a voltage on the gel, the proteins will start to move due to their negative electrical charge.
You must use suitable voltage, as the too high voltage will overheat the gel and maybe deform the bands.

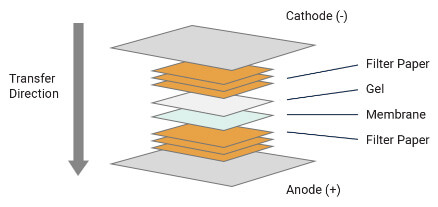
Transferring Proteins to A Membrane
Now you have finished the gel electrophoresis phase, and the following step is transferring the protein to the solid support membrane.
Your tools in this step will include sponges, filter papers, the gel, and the membrane, which is placed between the gel and the positive electrode.
In this process, you need to apply voltage to transfer the negatively charged proteins from the gel to the membrane.
The main types of membrane are:
- Nylon.
- Nitrocellulose.
- Polyvinylidene fluoride (PVDF).
The nylon membranes cause high background binding and irreversible staining of some dyes which makes them less common than the other two types of membrane, although they are superior in other several aspects.
The nitrocellulose membranes have a special advantage which is the low background regardless of the detection method.
But the nitrocellulose membranes should not be used for the transfer of proteins with low molecular weight, that is due to the relatively large average pore size,.
Furthermore, the membrane becomes brittle when they are dry, which makes them difficult to handle.
Regarding the PVDF membrane allows relabeling and it is more convenient to store due to its stability. In addition to the hydrophobic nature of PVDF provides high protein binding capacity, however, as a consequence, the background is also higher.
Regarding the methods of blotting you will have two methods:
- The wet method.
- The semi-dry method.
In the Semi-dry blotting less volume of buffer is needed and it is more rapid than the wet conditions.
This transfer method is usually less efficient, especially for larger proteins, and it may cause overheating and drying the gel when using extended transfer times.
At the same time, the wet method is preferred when the transfer must be efficient and give high quality regarding distinct and sharp bands.
Moreover, It is the most suitable choice for the transfer of larger protein complexes.
The gel, membrane, and filter papers are completely immersed in the buffer during the transfer and there is no risk of drying out the gel.
Pick The Best Virtual Plan for You

Blocking Nonspecific Sites
After the transferring of the proteins to membrane you need to prevent nonspecific binding of the antibodies to the membrane, rather than binding the protein of interest, a substance is used to block out the residual sites on the membrane. This is the fourth step, which is the antibody probing.
The substances that are commonly used are :
- Dried nonfat milk.
- 5% Bovine Serum Albumin (BSA) diluted in Tris Buffered Saline Tween (TBST).
- Normal goat serum.
- Casein.
- Fish gelatin.
Milk is inexpensive and easy to get hold of, however not suitable for all detection labels. And the fish gelatin can mask some proteins but it gives lower background as well as being a relatively expensive blocking buffer.
With regard to BSA, it is inexpensive, whereas serum can contain immunoglobulins giving rise to cross-reactivity.
Be careful during the selection of the blocking agent as it is considered as the key since none of the blocking buffers are ideal for all different antigen-antibody interactions.

The blocking procedure consists of incubating the membrane in the appropriate blocking buffer for an hour or longer.
You should perform the blocking procedure at +4°C if you are using long incubation times to rule out the risk of staining artifacts or background.
And take into consideration that you must achieve a delicate balance between reducing the background without decreasing the signal from the protein of interest.
The blocked membrane now is incubated with the primary antibody. The antibody is diluted to a suitable concentration in TBST, phosphate-buffered saline (PBS), or wash buffer. If the antibody is going to be reused, It is preferred to incubate the antibody with BSA.
Then after washing the membrane, the membrane will be incubated with the secondary antibody that binds to the primary antibody. The secondary antibody is labeled with a reporter.
When using a polyclonal antibody as a secondary antibody, it may give rise to some background.
In the case of background staining, the secondary antibody may be pre-blocked with non-immune serum from the host it was generated in.
Optimization of the concentration of the secondary antibody is recommended due to quite extended variations between antibodies as well as detection systems used.

Detection Methods
In the detection step, the protein-antibody-antibody complex is detected on the membrane.
There are various detection systems, based on chemiluminescence, chemifluorescence, fluorescence, chromogenic, or radioisotopic detection are available.
Amongst enzymes, the most common is HRP used together with chemiluminescent, chemifluorescent, or chromogenic substances. HRP has several advantages over other enzymes such as alkaline phosphatase (AP).
Also, it can be easily conjugated to antibodies or streptavidin which binds with extraordinarily high affinity to biotin, a commonly used tag, and can be used with different chemiluminescent reagents. In addition, it has a high substrate specificity giving a low background, stable, and inexpensive.
The most commonly used enzymatic detection system is chemiluminescence, based on antibodies conjugated to horseradish peroxidase (HRP).
The HRP enzyme catalyzes the oxidation of luminol from the luminol peroxide detection reagent. The multi-step reaction resulting in light emission.
You can use some chemicals like phenols that can enhance the emitted light.
A direct method is the use of fluorescence. Fluorescence-based detection systems use a fluorescent entity, or fluorophore, directly conjugated to an antibody or streptavidin.
The fluorophore can be excited using a light source of a specific wavelength causing light emission.
It is well suitable for quantitative western blot and since different fluorophores emit light of different wavelengths it is possible to perform multiplexing and specific detection of more than one protein at the time.
So the chemifluorescence is defined as using a chemical and/or an enzyme to induce the generation of an active fluorophore from a fluorogenic substrate.

Imaging
The last step in the Western blotting workflow before data analysis is image capture. You can perform the capturing step using a film, a CCD camera, or a scanner to collect the emitted light of the detection process.
Then the emitted light can be converted to an electrical signal using a photomultiplier tube (PMT) or a CCD camera. The electrical signal is then digitized for image display and analysis.

Analysis
The detected signals, using either X-ray film, scanners, or a charge-coupled device CCD camera, cause one or more visible protein bands on the membrane image.
To estimate the molecular weight of the protein you can make a comparison with marker proteins and the amount of protein can be determined as this is related to band intensity (within the limits of the detection system).
In most applications of western blotting , it is enough to confirm protein presence and roughly estimate the amount. However, other applications demand a quantitative analysis that defines protein levels in either relative or absolute terms.
For more information about Western blot analysis, please visit our blog “How to Analyze Western Blot Data“
What are the advantages of Western blotting?
Sensitivity: Because of its ability to detect as little as 0.1 nanograms of protein in a sample, the technique can theoretically serve as an effective early diagnostic tool, sensing even the slightest immunogenic response from a virus or bacteria in a patient sample.
Specificity: The western blot technique owes its specificity to two big contributing factors. First, gel electrophoresis sorts a sample into proteins of different size, charge, and conformation. This process in itself is a huge step towards detection, as bands formed in the gel already give clues about the size of the protein or polypeptide of interest.
The specificity of the antibody-antigen interaction serves as the second big factor. Because specific antibodies show affinity for specific proteins, the process can selectively detect a target protein even in a mixture of 300,000 different proteins.
Applications of Western Blotting
Western blot technique has a wide range of applications in scientific and clinical disciplines:
Western Blot Applications in Biological Sciences
A western blot is used to detect specific protein molecules from among a mixture of proteins. This mixture can include all of the proteins associated with a particular tissue or cell type.
Western blots can also be used to evaluate the size of a protein of interest and to measure the amount of protein expression. This procedure was named for its similarity to the previously invented method known as the Southern blot.
So we can summarize western blot uses as following :-
- Application of western blotting technique helps to identify the presence of a specific protein both by size and through the binding of an antibody makes them well-suited for evaluating levels of protein expression in cells, and for monitoring fractions during protein purification.

- Also, It is helpful for comparing expressions of a target protein from various tissues, or seeing how a particular protein responds to disease or drug treatment.
- In many cases, the western blot is used in combination with other key antibody-based detection techniques, such as immunohistochemistry.
Western Blot Applications in Medical
Western blots provide confirmation of results both in research and diagnostic testing.
In recent medical field, Western Blot has a wide range of applications in medical diagnosis, such as the application of medical diagnosis for HIV (Human Immunodeficiency Virus) infection, BSE (Bovine Spongiform Encephalopathy, also known as “mad cow disease”), FIV (Feline Immunodeficiency Virus), HBV (Hepatitis B Virus) infection, and so on. Usually, Western Blot is used as a confirmatory test for these diseases, following a high sensitivity ELISA (Enzyme-Linked Immuno Sorbent Assay) test with positive results.
For example, with HIV and prion disease, western blots are used as a key supplemental screen since their results are less ambiguous, and quicker than other methods.
What is the Western Blot test used for?
A Western blot test is typically used to confirm a positive HIV diagnosis. During the test, a small sample of blood is taken and it is used to detect HIV antibodies, not the HIV virus itself. The Western blot test separates the blood proteins and detects the specific proteins (called HIV antibodies) that indicate an HIV infection. The Western blot is used to confirm a positive ELISA, and the combined tests are 99.9% accurate.
Also, Western blot test is used to confirm or contest a diagnosis of HIV or Lyme disease after an ELISA antibody test comes back positive or negative. Since the ELISA test sometimes produces false positives, a second test is needed to further the diagnosis.
In case of Lyme disease, the Western blot test may detect antibodies to B. burgdorferi, the bacteria that causes this condition.
In case of HIV, the Western blot test may detect proteins from the envelope or core of the virus and enzymes generated by HIV infection.
PraxiLabs Virtual Labs include a range of 3D science experiments in physics, chemistry and biology experiments




